PointSuite
v.0.7
 |
PointSuite is a set of
programs to process macromolecular assemblies described by
point and helical symmetry operations, with the goals of
uniform annotation, archiving, and viewing. In order
to handle coordinates deposited in any orthogonal Cartesian
frame, the relationships between the deposition, standard
point and crystal frames are captured as frame
transformations. For example, the transformation
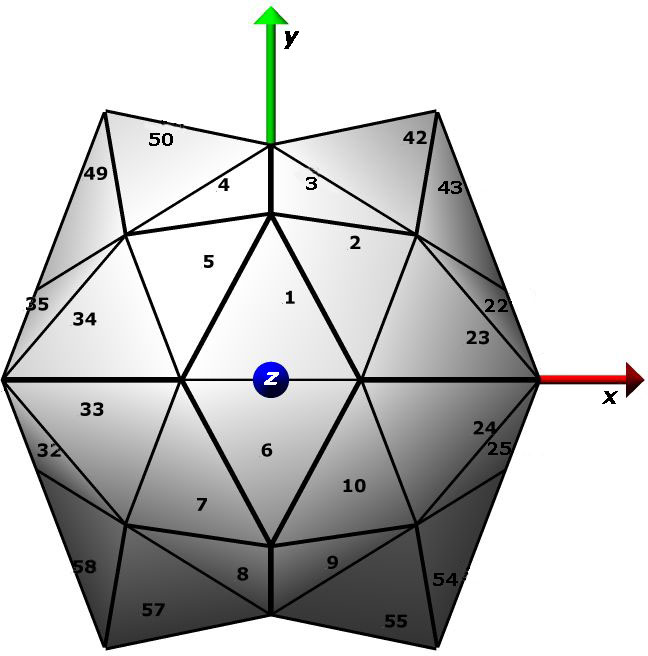
required to move icosahedral virus structures from deposited
position to the standard frame shown at left is calculated
and recorded. All point symmetries are fully handled;
helical entries are handled only for non-crystal cases.
Written/compiled by C. Lawson, with thanks to V.J. Reddy
(TSRI) for sharing PDB2VIPER code (findframe); Tom
Goddard (UCSF) for Chimera scripts (runchimera.csh);
Huanwang Yang (RCSB PDB) for importmats and cif-handling
subroutines.
Lawson CL, Dutta SD, Westbrook JD, Henrick K, Berman HM
(2008) Representation
of viruses in the remediated PDB archive, Acta Cryst D, 874-882.
|
DOWNLOAD: pointsuite-0.7.3.tgz
(version date: September 27, 2017)
|
Virus Processing Tutorial
|
|
Installation Instructions Documentation |
INSTALL/COMPILE/CONFIGURE:
Type the following commands in the directory where you want to
install the software.
tar xvzf
pointsuite0.7.tgz
cd pointsuite0.7
make (to compile)
The package is composed of programs written in C along with
C-shell-based scripts.
It has been extensively tested on linux and mac-intel osx
operating systems.
source
setup.csh
setup.csh script works for csh/tcsh shell. follow directions to
set up your environment more permanently.
To make
full use of the package, the graphics program UCSF Chimera should
also be installed and in your path.
TEST:
cd demo
rundemo.csh 1RUG (runs the 1RUG demo)
rundemo.csh all (runs all of the demos)
browse the demos to view functionality
1RUG: Generate archival cif for icosahedral virus crystal
structure.
1IFD: Generate archival cif for helical virus
fiber diffraction structure.
1EI7: Generate archival cif for D17 symmetry particle.
1CGM: Generate matrix representation for ~900 A length
helical TMV-like virus.
1M4X: Generate matrix representations for complex
virus particle sub-assemblies.
IMPORT: Generate BIOMT, CIF from typical
author-uploaded example input files using importmats.
Additional
icosahedral virus examples: 2XD8 (EM), 2W0C, 2VF9, 3N7X
(X-ray).
See Virus processing tutorial for more info.
RELEASE INFO:
version
0.5.8 (12 June 2007) initial stable release
version 0.6 (20 June 2011) minor updates:
*importmats (from H. Yang) handles additional matrix type
(xncsrel) from CNS ncs.def.
*update of scripts automating image generation to work with v.1.4
Chimera and higher
*when run without arguments, runpt.csh autoscript now prints brief
documentation
*new
virus processing tutorial
*additional
documentation now provided for utilities: importmats, autoscripts,
multiplymats
*RCSBvirusimages.csh script to generate set of virus images for
web display.
version 0.7 (15 January 2013):
*improved cif parsing subroutines added by H. Yang (cifparse.c).
*file input reading improvements in importmats, findframe,
makeassembly
*findframe single input file with matrices and coordinates can now
be either PDB or CIF; optional 2nd file in BIOMT format (overrides 1st file matrices)
*new program cif2pdb creates simple pdb file (matrices, cryst1
record, coordinates) from cif (H. Yang).
*simplified scripts, PDB-dependency removed for runpt.csh
*RCSBvirusimages.csh script handles split entry cases
(modifications by Ezra Peisach)
0.7.3 release (27 September 2017) includes updates to importmats (modifications by Huanwang Yang)
PROGRAM
DOCUMENTATION:
PROGRAM FINDFRAME
Description
The program FINDFRAME calculates the transformation matrix that
moves the asymmetric unit of a particle with point symmetry in an
arbitrary (skew) frame into a defined position within a standard
point frame. The standard icosahedral
frame is defined as having the 5-fold axis of the first pentamer
centered on the vector (x=0,y=1, z=phi), where phi is the
golden ratio (sqrt(5)+1)/2. This convention is also employed
by the VIPER database and the is proposed convention of Belnap et.
al. for cryoEM maps.
FINDFRAME is an extension of the PDB2VIPER
program from the VIPERdb (Shepherd, et al. (2006) Nucleic Acids
Res, 34, D386-389) that incorporates the qikfit least-squares
fitting routine from Bioplib
(A.C.R. Martin, personal communication). Algorithm steps
have been added to increase the precision of the calculated
transformation matrix and to improve uniformity of coordinate
placement relative to the standard icosahedron symmetry axes (see
description below).
Use
on the command line:
>findframe
infile.pdb/cif
The input file is expected to have all needed transformation
matrices to build the icosahedral or other point group particle
AND coordinates for the asymmetric unit . PDB
matrices can be given either as REMARK 350 BIOMT or MTRIX records;
in cif the matrices are given in _pdbx_struct_oper_list and must
have type of "general operation", "point symmetry operation" or
"helical symmetry operation". One of
the transformation matrices must be the identity element. Optional:
a second file can be provided with BIOMT records; in this case the
matrices in the 2nd file override any present in the first file.
Algorithm
modifications to PDB2VIPER (from V.J. Reddy)
are in italics
- The approximate center of mass
("reference atom") is calculated from Calpha and P atom
positions.
- The set of matrix translation
vectors are averaged to find the center of the particle relative
to the coordinate origin. The reference atom is translated by
the negated particle center and transformed into a unit vector.
- The rotation matrices are
decomposed into angle-axis form. The angle identifies the fold
of the rotation (e.g. 72 or 144 degrees for 5-fold, 120 degrees
for 3-fold). The axis vector defines the symmetry orientation.
- The rotations are
checked against their corresponding translation vectors to
identify helical symmetry. If helical symmetry is
detected, then the program attempts to identify all of the
helical parameters from the matrices, and will print this info
to findframe.cif and will then exit (currently, program only
handles cases with helical axis on z).
- If no helical operations are
detected, then point symmetry is assumed and the program
analyses the matrices to deduce the correct symmetry. The
remainder of the algorithm description explains what happens for
icosahedral symmetry, but essentially similar steps are taken
for the other symmetries (circular,dihedral, tetrahedral,
octahedral).
- The five-fold, two-fold, and
three-fold closest to the reference atom are identified.
- The position of the reference
atom unit vector relative to the 5-3 and 5-2 planes is
evaluated. If the reference atom
is closer to the 5-3 plane, the icosahedral a.u. is classified
as "3-fold centric" and the closest 3-fold is selected for
alignment in the next step. If
the reference atom is closer to the 5-2 plane (typically true
for T=3 viruses), the icosahedral a.u. is classified as
"2-fold centric" and the 3-fold to the right of the 2-fold is
selected for alignment.
- The input structure's 5-fold
and 3-fold are aligned onto the standard icosahedral frame
5-fold (0, 1, phi) and 3-fold (phi/3,
0, (2*phi+1)/3)
axes
in
two
steps.
First,
the
rotation
that
superimposes the normal to the 5-3 plane of the deposited
structure onto the normal to the 5-3 plane of the standard
icosahedron 5-fold is found and applied. Second,
the rotation around the aligned normals that superimposes the
pair of 5-fold axes is found and applied. The
initial estimate for the findframe matrix is based on these two
rotation matrices and the translation vector determined in step
2.
- Fitting/refinement: The
input-supplied transformations are applied to the reference
atom, and the resulting 60-atom constellation is transformed
to the icosahedral frame by the initial findframe matrix. A reference set is generated by
applying icosahedral symmetry operations to the 1st transformed atom.
The 60-atom constellation is fitted to the icosahedral
reference set, yielding a correction matrix. The final
findframe matrix is generated by applying the correction
matrix to the initial estimate. This
step is particularly helpful for cases where the input
file-supplied matrices have either low precision or small
random errors, as these errors tend to be averaged out.
Output
General info about the calculation is provided
in the standard output. On successful execution a cif file
with symmetry and frame transformation info is also output
called *findframe.cif*.
PROGRAM POINTMATS
Description
Generates sets of transformations corresponding
to point or helical symmetry provided in the input cif file (
_pdbx_point_symmetry, _pdbx_helical_symmetry).
If the input cif contains a "transform to point frame"
matrix with _pdbx_struct_oper_list.id labelled 'P' (or a
"transform to helical frame" matrix is given with
_pdbx_struct_oper_list.id labelled 'H'), the matrix set is
transformed such that it can be applied to coordinates away from
the standard frame, e.g., [P-inv][std mats][P]. Use
pointmats to obtain simple matrix set output files for
point/helical symmetry operations; use makeassembly if you need
full assembly and asymmetric unit descriptions.
Use
Commonly used after findframe, e.g.:
pointmats findframe.cif
will generate point or helical matrix set with standard order in the
same frame as the matrices analysed by findframe.
Output
General info about the calculation is provided
in the standard output; the matrices are written in CIF format to
pointmats.cif and in BIOMT format to pointmats.biomt. Point
symmetry operations follow a standard order. Helical
symmetry operations are given as a continuous run centered around
the identity element.
PROGRAM
MAKEASSEMBLY
Description
The set of point symmetry operations
corresponding to a crystal asymmetric unit is identified, given
the following input: CIF file with unitcell, spacegroup, asym_id,
entity_id records , cif with _pdbx_point _symmetry or
_pdbx_helical_symmetry and _pdbx_struct_oper_list with frame
transformations (P for "transform to point frame" ; H for
"transform to helical frame"; X0, X1, etc. for "transform to
crystal frame"). The program will analyse the structure for
ncs only if at least one "transform to crystal frame" matrix is
given (X0) (this will frequently be the identity matrix). FROM
v.5.7 onward, makeassembly outputs asym_id lists instead of
author_asym id list.
Use
Crystal frame transformations are optional (e.g., for EM
structures):
>makeassembly uc_symtry_scale_.cif
symm_transforms.cif
Algorithm
- For each independent particle
position n defined by Xn, the full standard set of matrices for
the point symmetry given in 2ptmat , **to be applied to
coordinates transformed by X0**, are calculated as
[Xn][2ptmat^-1][StdMats][2ptmat][X0^-1].
- The crystal symmetry matrices in
their fractional forms are transformed by the translation part
of [Xn^-1]. This moves the
origin of the crystal lattice to the particle center. Crystal symmetry operators
passing through the particle center are identified by lack of
fractional translation components (full unit translations are
reset to 0).
- Each crystal symmetry rotation
identified in step 2 is applied to each of the rotations
calculated in step 1. Symmetry-transformed
rotations that are identical to untransformed rotations with
lower index are eliminated from the list of ncs operations.
Output
General info about the calculation for each
independent particle is provided in the standard output; an
archival cif (assembly.cif) is generated, as well as biomt records
for the full assembly (assembly.biomt). For crystal
structures a bare-bones crystal frame pdb file is created that can
be input to sfcheck or packing programs (assembly_xframe.pdb), and
ncs records are generated (assembly.ncs).
Utilities
importmats:
call with
>importmats matfile
Reads in and automatically detects a wide variety of matrix
record formats including BIOMT, MTRIX, ncs.def.
Outputs file with BIOMT records named "importmats.biomt" and file
with CIF _pdbx_struct_assembly_oper records named "importmats.cif"
Use importmats to prepare author.biomt file for runpt.csh
autoscripts:
(1) make assembly cif records:
runpt.csh (prints out instructions for preparing script arguments)
runpt.csh entry.cif author-upload-matfile (noncrystal and most
crystal structures)
runpt.csh entry.cif author-upload-matfile X0.mat (crystal out of
frame or multiple positions in crystal a.u.)
(2) makes pictures of files generated by runpt.csh:
runchimera.csh
(3) makes pictures from PDB file ready for release:
RCSBvirusimages.csh file1.pdb
RCSBvirusimages-split.csh file1.pdb file2.pdb file3.pdb ...
(combined images for split entries)
frac2orth:
call with
>frac2orth
Interactively requests 6 parameters of unit cell and a position in
fractional coordinates, outputs orthogonalization matrix,
fractionalization matrix, and corresponding Cartesian coordinates of
input position. Useful if the translation part of a skew
matrix is provided in fractional coordinates.
movecoords:
call with
>movecoords file1.pdb file2.matrix
reads in pdb file and 4x4 matrix file, writes out "new.pdb" file
that is identical to input file except with x,y,z coordinates
tranformed by the 4x4 matrix.
multiplymats:
Performs complex matrix multiplications
given a list of matrices in cif format and a string defining the
desired multiplication.
String can include numerical ranges and recursion.
examples:
"(1-2)(3,5,7-9)" creates the set of matrices 1*3, 1*5,
1*7, 1*8, 1*9, 2*3, 2*5, 2*7, 2*8, 2*9
"(1)(2,(2)(2),(2)(2)(2))" creates the set of matrices 1*2,
1*2*2, 1*2*2*2
call with
>multiplymats <pointmats or assembly cif> <matrix
multiplication string in double quotes>
example (1m4x):
multiplymats 1m4x.cif "(1-60)(61-88)"
number of matrices read: 89
Matrix multiplication expression to be parsed: (1-60)(61-88)
result:
1*61
1*62
1*63
1*64
1*65
1*66
1*67
1*68
1*69
1*70
1*71
...
60*85
60*86
60*87
60*88
Writing 1680 matrices to *mult.cif*
Writing 1680 matrices to *mult.biomt*
last modified 14 Jan 2013
C. Lawson